MDR Transition…a compliant approach to current Medical Device Regulation (EU MDR 2017/745)
Change is always hard to accept. However, experiencing the EU MDR transition from MDD (Medical Device Directive) in The Supplier Quality department, was not so challenging after all. At the time of starting the transition it did appear to be a daunting task. However, by separating all new clauses and expectations in the MDR, the work effectively divided and managed amongst the workstreams.
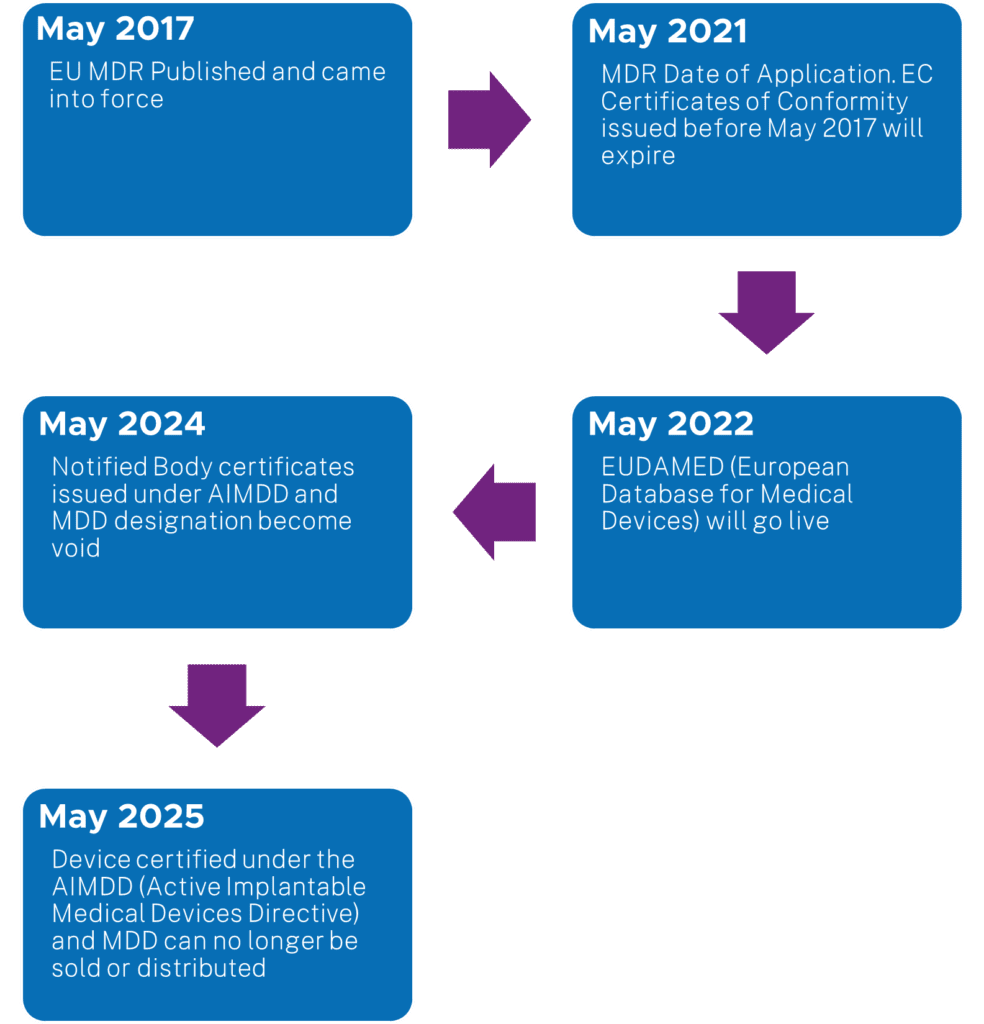
Before sharing information on the approach method for this project, below is a snapshot chart for the EU MDR timelines:
Having extensively familiarised with the new MDR, the key changes that should be considered within the supply chain for devices, are:
- CE marking shall be affixed on devices complying with the MDR. Devices will freely circulate in the European Economic Area and Switzerland provided that the manufacturer will continue complying with their obligations under the MDR and that the devices are safe.
- The traceability of devices by means of a Unique Device Identification system (UDI system) based on international guidance should significantly enhance the effectiveness of the post-market safety-related activities for devices due to:
- improved incident reporting
- targeted field safety corrective actions
- better monitoring by competent authorities
- should also help to reduce medical errors
- and aid in the fight against falsified devices.
Use of the UDI system should also improve purchasing and waste disposal policies and stock-management by health institutions and other economic operators and, where possible, be compatible with other authentication systems already in place in those settings.
- As per article 33, EUDAMED shall include the following electronic systems:
- the electronic system for registration of devices
- the UDI-database
- the electronic system on registration of economic operators
- the electronic system on notified bodies and on certificates
- the electronic system on clinical investigations
- the electronic system on vigilance and post-market surveillance
- the electronic system on market surveillance.
- Identification within the Supply Chain: Distributors and importers shall co-operate with manufacturers or authorised representatives to achieve an appropriate level of traceability of devices. Economic operators shall be able to identify the following:
- any economic operator to whom they have directly supplied a device
- any economic operator who has directly supplied them with a device
- any health institution or healthcare professional to which they have directly supplied a device.
From having taken an organisation’s supplier qualification team through this transition, the below implementation approach would be recommended. This project was focused on Distribution of Medical Device and on the supply chain route compliance in accordance with EUMDR.
- Complete Gap assessment: Using our gap assessment tool, each of the new sections of EUMDR, which applies to Distributors, were verified against the current operations of company documents and policies.
- Change control: This was initiated to cover gaps in the QMS to update including policies and procedures.
- Identification and delegation of work: On completion of gap assessment, the approach was taken to delegate work within a cross functional team to complete updates on the applicable documents.
- Update of Level 1 and Level 2 documents: e.g., Site Master file, Standard Operating Procedures, Forms, Templates. This work was completed with support from interdepartmental colleagues, closely work with Quality Assurance.
- Training: On completion of the updates, training was arranged for employees that needed to understand the changes to the existing processes and how it will affect the daily tasks.
To summarise on the high-level changes in the EUMDR 2017/745, these relate to CE Marking, EUDAMED Database, Implementation of UDI (Unique Device Identifier), Authorised Representative and Economic Operator.
Written by: Dharmapal Jadeja, GMP/GDP QA Manager